Piotr Piecuch

Research
Quantum Chemistry and Physics
(Research Description PDF)
My research program focuses on (i) ab initio quantum theory of molecular electronic structure and other many-body systems, (ii) molecular properties, spectroscopy, and photochemistry, (iii) reaction mechanisms and dynamics, and (iv) theory of intermolecular forces. We design and apply quantum-mechanical methods that enable precise determination of potential energy surfaces and property functions for both existing and hypothetical molecular species in their ground and excited states. We are also interested in accurate quantum calculations for strongly correlated systems, weakly interacting molecular clusters, and atomic nuclei.
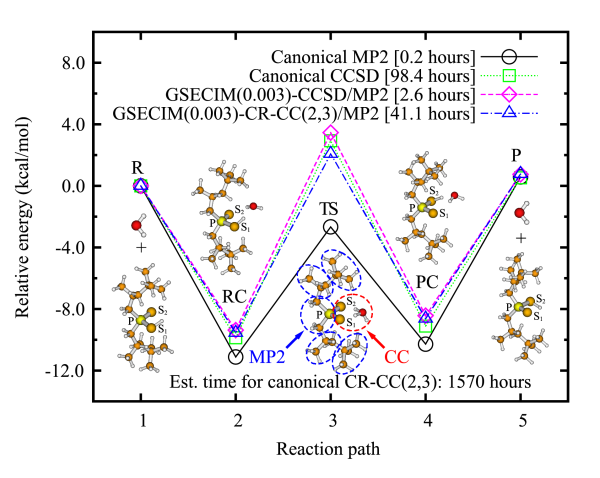
Quantum theory of molecular electronic structure. The key to understanding molecular electronic structure and dynamical behavior of molecules is an accurate assessment of the many-electron correlation effects. Our group focuses on the development and applications of new quantum-mechanical methods that include correlation, particularly on the coupled-cluster theory and its renormalized, active-space, extended, multi-reference, and response variants that allow us to study bond breaking, electronically excited states, electron-transfer processes, molecular properties in vibrationally and electronically excited states, and transition probability coefficients for various types of spectroscopy. We examine ways of achieving high-level coupled-cluster or numerically exact energetics by combining deterministic computations with stochastic wave function sampling. We also develop approximate coupled-pair approaches for strongly correlated systems and local correlation coupled-cluster methods and their multi-level extensions that can be applied to high accuracy ab initio calculations for systems with hundreds of atoms. Our primary interest is in high-accuracy methods that allow us to be predictive. We write computer codes for the standard and new coupled-cluster methods which are distributed world-wide through a popular electronic structure package GAMESS and plugins to PSI4 available on GitHub. Some of our methods are also available in NWChem, Q-Chem, and MRCC packages.
Many-body methods of quantum mechanics and nuclear physics. We demonstrated that quantum-chemistry-inspired coupled-cluster methods can be applied to atomic nuclei. We performed several highly successful ab initio coupled-cluster calculations for 4He, 16O, and valence systems around 16O using modern nucleon-nucleon interactions. We also carried out widely publicized coupled-cluster calculations for 56Ni and its isotopes. We are looking for the alternative approaches to accurate calculations for many-fermion systems with pair-wise interactions, including the use of cluster expansions involving two-body correlation operators to represent nearly exact many-fermion states.
Molecular properties, spectroscopy, and photochemistry. We use linear-response coupled-cluster methods, along with other ab initio approaches, to calculate molecular multipole moments and (hyper)polarizabilities and the effect of nuclear motion on these properties. We use first-principles theories to obtain rovibrational, electronic, and rovibronic spectra of molecules and weakly bound species. We have demonstrated that the lowest excited state of methylcobalamin should be interpreted as metal-to-ligand charge-transfer excitation and that azulene possesses the doubly excited state below the ionization threshold, which can drive multi-photon ionization experiments related to Rydberg fingerprint spectroscopy. We have provided definitive information about structural, electronic, and spectroscopic properties of several organic biradicals and small metal nanoparticles, including, for example, beryllium, magnesium, silver, and gold clusters.
Reaction mechanisms and dynamics. We performed successful computational studies for several important organic chemistry reactions, including the Cope rearrangement of 1,5-hexadiene, cycloaddition of cyclopentyne to ethylene, thermal stereomutations of cyclopropane, and isomerization of bicyclo [1.1.0] butane to buta-1,3-diene. We carried out unprecedented coupled-cluster calculations for CuO2 and Cu2O2 systems, relevant to oxygen activation by metalloenzymes, for photoisomerizations of acetylacetone, for diffusion of atomic oxygen on the silicon surface, for proton-transfer reactions between the dithiophosphinic acids and water molecules, for aerobic oxidation of methanol on gold nanoparticles, and for the Co-C bond dissociation in methylcobalamin, relevant to catalytic properties of B12. We also studied the photo-induced charge-transfer (“harpooning”) reactions between alkali and alkaline earth metal atoms and halides. In particular, we combined ab initio and dynamical approaches to characterize quasi-bound states of van der Waals molecules that are precursors of these reactions.
Intermolecular interactions. Intermolecular potentials are a necessary ingredient for the determination of the structure, stability, and dynamics of weakly bound clusters and condensed phases. We are interested in many-body interactions, which are important when three or more atoms or molecules interact, and study interactions in dimers.
Contact / Webpage
Area(s) of Interest
Theoretical and Computational (Th)
Chemical Physics (CP)
Inorganic (In)
Nuclear (Nu)
Organic (Or)
Organometallic (Om)
Physical (Ph)
Selected Publications
Quantum Computation Solves a Half-Century-Old Enigma: Elusive Vibrational States of Magnesium Dimer Found, S.H. Yuwono, I. Magoulas, and P. Piecuch, Sci. Adv. 2020, 6, eaay4058.
Recent Developments in the General Atomic and Molecular Electronic Structure System, G.M.J. Barca et al., J. Chem. Phys. 2020, 152, 154102.
Steric Effects in Light-Induced Solvent Proton Abstraction, J. Lahiri et al., Phys. Chem. Chem. Phys. 2020, 22, 19613.
Accelerating Convergence of Equation-of-Motion Coupled-Cluster Computations Using the Semi- Stochastic CC(P;Q) Formalism, S.H. Yuwono, A. Chakraborty, J.E. Deustua, J. Shen, and P. Piecuch, Mol. Phys. 2020, 118, e1817592.
Photoluminescence, Photophysics, and Photochemistry of the VB— Defect in Hexagonal Boron Nitride, J.R. Reimers, J. Shen, M. Kianinia, C. Bradac, I. Aharonovich, M.J. Ford, and P. Piecuch, Phys. Rev. B 2020, 102, 144105.
The Ground State Electronic Energy of Benzene, J.J. Eriksen et al., J. Phys. Chem. Lett. 2020, 11, 8922.
Isoenergetic Two-Photon Excitation Enhances Solvent-to-Solute Excited-State Proton Transfer, J. Lahiri et al., J. Chem. Phys. 2020, 153, 224301.
High-Level Coupled-Cluster Energetics by Monte Carlo Sampling and Moment Expansions: Further Details and Comparisons, J.E. Deustua, J. Shen, and P. Piecuch, J. Chem. Phys. 2021, 154, 124103.
CV
M.Sc., 1983, Univ. of Wroclaw (Poland)
Ph.D., 1988, Univ. of Wroclaw
Postdoctoral Fellow, 1988-91, Univ. of Waterloo (Canada)
Assistant Professor, 1990-92, Univ. of Wroclaw
Postdoctoral Associate, 1992-93, Univ. of Arizona
Visiting Assistant Professor, 1994-95, Univ. of Waterloo
Visiting Assistant Professor, 1995-97, Univ. of Toronto
Postdoctoral Associate, 1997-98, Univ. of Florida
Adjunct Assistant Professor, 2000-2003, Univ. of Waterloo
Alfred P. Sloan Research Fellow, 2002-04
Elected Member, Eur. Acad. of Arts, Sciences and Humanities, Paris, France, 2003
Invited Fellow of JSPS and Vis. Prof., 2005, Kyoto Univ. (Japan)
Vis. Prof., Univ. of Coimbra (Portugal), 2006
Palit Memorial Lecture, IACS, Kolkata, 2007
Fellow of the American Physical Society, 2008
Fellow of the American Association for the Advancement of Science, 2011
Invited Prof., Inst. for Molecular Sci., Okazaki (Japan), 2012-13
Distinguished Fellow, Kosciuszko Foundation Collegium of Eminent Scientists, 2015
Clark Way Harrison Distinguished Visiting Professor, Washington Univ. in St. Louis, 2016
Fellow of the Royal Society of Chemistry, 2016
Schaad Lectureship, Vanderbilt Univ., 2017
Elected Member, International Academy of Quantum Molecular Science, 2018
Awards
| Year | Award | Organization |
|---|---|---|
| 2020 | MSU Foundation Professor | Michigan State University |
| 2019 | The Xingda Lectureship | Peking University |
| 2018 | Elected Member | International Academy of Quantum Molecular Science |
| 2018 | International Academy of Quantum Molecular Science | |
| 2017 | The Lawrence J. Schaad Lectureship in Theoretical Chemistry | Vanderbilt University |
| 2017 | Co-organizer | 49th Midwest Theoretical Chemistry Conference, East Lansing, Michigan |
| 2016 | Fellow | Royal Society of Chemistry |
| 2016 | Co-organizer | Symposium on Electronic Structure Theory "Advances in Electron Correlation: From Strongly Correlated to Large Systems" at the 9th Congress of the International Society for Theoretical Chemical Physics, Grand Forks, North Dakota |
| 2016 | Elected Fellow | Royal Society of Chemistry |
| 2016 | Clark Way Harrison Distinguished Visiting Professor | Washington University in St. Louis |
| 2015 | Co-organizer | Symposium “Recent Progress in Molecular Theory for Excited-State Electronic Structure and Dynamics” at Pacifichem 2015 |
| 2015 | Distinguished Fellow of the Collegium of Eminent Scientists | The Kosciuszko Foundation |
| 2014 | Adjunct Professor | Michigan State University |
| 2014 | Outstanding Reviewer | Chemical Physics Letters (Elsevier publication) |
| 2013 | International Advisory Board | 8th Congress of the International Society for Theoretical Chemical Physics, Budapest, Hungary |
| 2013 | International Scientific Committee | 18th International Workshop Quantum Systems in Chemistry and Physics, Paraty (Rio de Janeiro), Brazil |
| 2012 | Co-editor | Progress in Theoretical Chemistry and Physics, Volume 22 (2012) |
| 2012 - 2013 | Invited Professor and Scientist | Institute for Molecular Science, Okazaki, Japan |
| 2012 | International Scientific Committee | 17th International Workshop Quantum Systems in Chemistry and Physics, Turku, Finland |
| 2012 | Co-editor | Progress in Theoretical Chemistry and Physics, Volume 26 (2012) |
| 2011 | International Scientific Committee | 7th Congress of the International Society for Theoretical Chemical Physics, Tokyo, Japan |
| 2011 | Elected Fellow | American Association for the Advancement of Science |
| 2011 | International Scientific Committee | 16th International Workshop Quantum Systems in Chemistry and Physics, Kanazawa, Japan |
| 2011 | Guest Co-editor | International Journal of Quantum Chemistry, Volume 111, Issue 2 (2011) |
| 2010 | International Scientific Committee | 15th International Workshop Quantum Systems in Chemistry and Physics, Cambridge, UK |
| 2010 | Editorial Board | Handbook of Research on Computional and System Biology: Interdisciplinary Applications |
| 2009 | Editorial Board | Research Letters in Physical Chemistry |
| 2009 | International Scientific Committee | 14th International Workshop Quantum Systems in Chemistry and Physics, San Lorenzo de El Escorial, Madrid, Spain |
| 2009 | Editorial Board | Advances in Physical Chemistry |
| 2009 | Co-editor | Progress in Theoretical Chemistry and Physics, Volume 19 (2009) |
| 2009 | Co-editor | Progress in Theoretical Chemistry and Physics, Volume 20 (2009) |
| 2008 | Elected Fellow | American Physical Society |
| 2008 | Co-organizer | 6th Congress of the International Society for Theoretical Chemical Physics, Vancouver, Canada |
| 2008 | Organizer | 13th International Workshop Quantum Systems in Chemistry and Physics, Lansing, Michigan |
| 2008 | Co-editor | AIP Conference Proceedings, Volume 995 |
| 2008 | Co-editor | Progress in Theoretical Chemistry and Physics, Volume 18 (2008) |
| 2008 | Editorial Board | Interdisciplinary Sciences: Computional Life Sciences |
| 2008 | International Scientific Committee | 13th International Workshop Quantum Systems in Chemistry and Physics, Lansing, Michigan |
| 2008 | International Scientific Committee | 6th Congress of the International Society for Theoretical Chemical Physics, Vancouver, Canada |
| 2007 - 2009 | Editorial Board | Open Chemical Physics Reviews |
| 2007 | International Scientific Committee | 12th International Workshop Quantum Systems in Chemistry and Physics, London, UK |
| 2007 | Editorial Board | The Open Chemical Physics Journal |
| 2007 | Editorial Board | Progress in Theoretical Chemistry and Physics |
| 2007 | Guest Co-editor | International Journal of Quantum Chemistry, Volume 107, Issue 14 (2007) |
| 2007 | International Advisory Committee | 9th Conference Molecular Spectroscopy, Wroclaw-Ladek-Zdroj, Poland |
| 2007 | Co-organizer | 2nd Workshop on "Nuclei and Mesoscopic Physics", East Lansing, Michigan |
| 2007 | The S.R. Palit Memorial Lecture | Indian Association for the Cultivation of Science, Kolkata, India |
| 2007 | University Distinguished Professor | Michigan State University (Department of Chemistry) |
| 2006 | Visiting Professor | University of Coimbra, Portugal (Department of Chemistry) |
| 2006 | International Scientific Committee | 11th International Workshop Quantum Systems in Chemistry and Physics, St. Petersburg, Russia |
| 2005 | Editorial Board | International Journal of Quantum Chemistry |
| 2005 | Editorial Board | Journal of Computational Methods in Science and Engineering |
| 2005 | Invitation Fellowship and Visiting Professorship | Japan Society for the Promotion of Science, Kyoto University, Japan |
| 2004 | Organizer | 36th Midwest Theoretical Chemistry Conference, East Lansing, Michigan |
| 2004 | QSCP Promising Scientist Prize | Centre de Mecanique Ondulatoire Appliquee, France |
| 2004 | Co-organizer | Workshop "Nuclei and Mesoscopic Physics", East Lansing, Michigan |
| 2004 | Professor | Michigan State University (Department of Chemistry) |
| 2004 - 2010 | Adjunct Professor | Michigan State University (Department of Physics and Astronomy) |
| 2003 | Elected Corresponding Member | European Academy of Sciences, Arts, and Humanities, France |
| 2003 | Organizer | Workshop "High-Performance Computer Center at Michigan State University" |
| 2003 - 2004 | Adjunct Associate Professor | Michigan State University (Department of Physics and Astronomy) |
| 2002 - 2004 | Alfred P. Sloan Research Fellowship | Alfred P. Sloan Foundation |
| 2002 - 2004 | Associate Professor | Michigan State University (Department of Chemistry) |
| 2000 | Young Investigator Award | Wiley-International Journal of Quantum Chemistry |
| 2000 - 2003 | Adjunct Assistant Professor | University of Waterloo, Canada (Department of Applied Mathematics) |
| 1998 - 2002 | Assistant Professor | Michigan State University (Department of Chemistry) |
| 1997 - 1998 | Postdoctoral Research Associate | University of Florida (Department of Chemistry (QTP)) |
| 1995 - 1997 | Visiting Assistant Professor | University of Toronto, Canada (Department of Chemistry) |
| 1994 - 1995 | Visiting Assistant Professor | University of Waterloo, Canada (Department of Applied Mathematics) |
| 1992 | Award for Research | The Polish Chemical Society |
| 1992 - 1993 | Postdoctoral Research Associate | University of Arizona (Department of Chemistry) |
| 1991 | The President of the University of Wroclaw Award for Research | University of Wroclaw, Poland |
| 1989 | Award for Outstanding Doctoral Dissertation | The Minister of National Education of Poland |
| 1988 | Ph.D. | University of Wroclaw, Poland (Faculty of Chemistry) |
| 1988 - 1991 | Postdoctoral Fellow | University of Waterloo, Canada (Department of Applied Mathematics) |
| 1987 | The President of the University of Wroclaw Award for Research | University of Wroclaw, Poland |
| 1986 | Award for Research | The Polish Chemical Society |
| 1986 | The President of the University of Wroclaw Award for Research | University of Wroclaw, Poland |
| 1985 | The President of the University of Wroclaw Award for Research | University of Wroclaw, Poland |
| 1983 | Award for the Best Master of Science Dissertation in Poland in Academic Year 1982-83 | The Polish Chemical Society |
| 1983 | M.S. | University of Wroclaw, Poland (Faculty of Chemistry) |
| 1983 | The President of the University of Wroclaw Award for Research | University of Wroclaw, Poland |
| 1982 | Award for Undergraduate Research in Chemistry (twice) | The Polish Academy of Sciences |