Robert Cukier

Research
Theory and Computation of Protein Structure Function Relations
(Research Description PDF)
Statistical Mechanics and Quantum Mechanics are themethods we use to create theories and computa- tional algorithms for the simulation of protein structure-function relations. A major effort is devoted to accelerating MD to reach realistic time scales. Another area of interest is ab initio molecular dynamics of electron localization in a variety of liquids. Ongoing studies include simulations and analysis of large-scale protein domain movements, proton translocation, and excess electron localization.
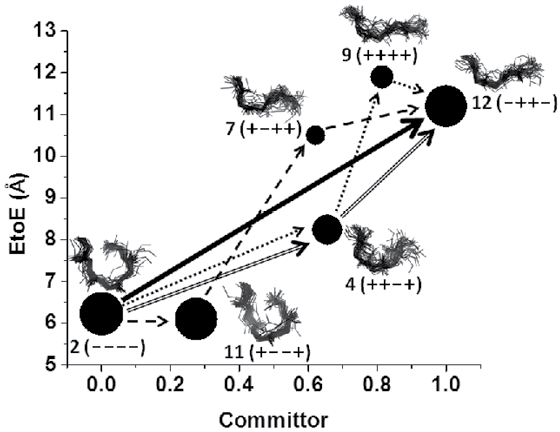
New methods are under development that can enhance the sampling of protein configurations to be able to explore the free energy cost for large domain motions. In the five residue opioid peptide met- enkephalin we have used long simulations along with a clustering and transition path analysis to obtain the major pathways of dihedral states visited for transitions between open and closed configurations and their correlation with committors (small values mean intermediate states return to initial state before visiting final state and vice versa).
Protein stability is based on a delicate balance between energetic and entropic factors. Intrinsically disordered proteins (IDPs) interacting with a folded partner protein in the act of binding can order the IDP to form the correct functional interface by decrease in the overall free energy. We develop methods evaluate the part of the entropic cost of ordering an IDP arising from their dihedral states. As shown in the plot below, the conformations based on the dihedral sampling is very dependent among all the dihedrals. This leads to a reduction of the entropic cost of binding relative to independent dihedral conformations.

Area(s) of Interest
Theoretical and Computational (Th)
Biological (Bi)
Physical (Ph)
Selected Publications
Dihedral Angle Entropy Measures for Intrinsically Disordered Proteins, Cukier R. I., J. Phys. Chem. B 2015, 119, 3621−3634.
Conformational Transition of Response Regulator RR468 in a Two-Component System Signal Transduction Process, Banerjee R. Yan H, and Cukier R. I., J. Phys. Chem. B 2014, 118, 4727-4742.
Transition Paths of Met-Enkephalin from Markov State Modeling of a Molecular Dynamics Trajectory, Banerjee R. and Cukier R. I., J. Phys. Chem. B 2014, 118, 2883-2895.
Computational Simulation Strategies for Analysis of Multisubunit RNA Polymerases, Wang B. B., Feig M., Cukier R. I., & Burton Z. F., Chem. Revs. 2013, 113, 8546-8566.
Excess Dielectron in an Ionic Liquid as a Dynamic Bipolaron, Liu J. X., Wang Z. P., Zhang M., Cukier R. I., and Bu Y. X., Phys. Rev. Letts. 2013, 110, 107602.
Variance of a Potential of Mean Force Obtained Using the Weighted Histogram Analysis Method, Cukier R. I., J. Phys. Chem. B 2013, 117, 14785-14796.
Simulations of Temperature and Salt Concentration Effects on bZIP, a Basic Region Leucine Zipper, Cukier R. I., J. Phys. Chem. B 2012, 116, 6071-6086.
A Hamiltonian replica exchange method for building protein-protein interfaces applied to a leucine zipper, Cukier R. I., J. Chem. Phys. 2011, 134, 045104.
CV
B.A., 1965, Harpur College
M.S., 1967 and Ph.D., 1969, Princeton Univ.
NATO Fellow, 1969, Lorentz Institute for Theoretical Physics
Research Associate, 1971, Univ. of California-San Diego
Alfred P. Sloan Research Fellow, 1976-80
Guggenheim Memorial Fellow, 1979-80
Co-Director: Quantitative Biology and Modeling Initiative
Awards/Honors
| Year | Award | Organization |
|---|---|---|
| 1979 - 1980 | Fellow | John Simon Guggenheim Foundation |
| 1976 - 1980 | Alfred P. Sloan Fellowship | Alfred P. Sloan Foundation |
| 1969 | Ph.D. | Princeton University |
| 1969 - 1971 | Postdoctoral Fellow | NATO |
| 1967 | NBS Summer Fellow | National Bureau of Standards |
| 1967 | M.S. | Princeton University |
| 1967 - 1969 | NIH Predoctoral Fellow | National Institutes of Health |
| 1965 - 1966 | NSF Trainee | National Science Foundation |
| 1965 | Bachelor of Arts | Harpur College |